
本文来自微信公众号: 范阳 ,编辑:范阳,作者:范阳
前言
Intro
今年人工智能领域最大的趋势之一,便是前沿实验室(frontier labs)在由外部合作伙伴生成的专属训练数据上投入了极其惊人的资金。虽然这些实验室的支出指标极少公开披露,但可以通过数据标注公司自身的数据来进行估算。早在2025年6月,该领域最大的两家巨头—Surge AI和Scale AI的平均年营收就已分别达到了约10亿美元,而这主要正是由前沿实验室的疯狂砸钱所驱动的。
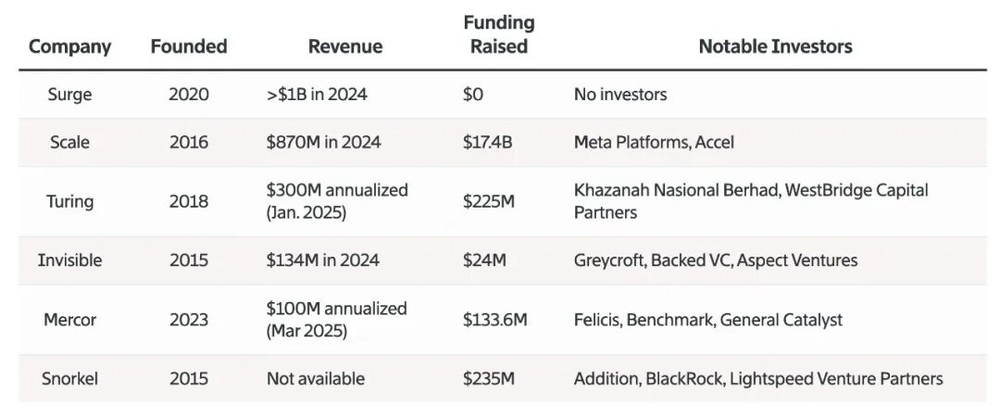
顶级数据标注公司的收入。(数据来源于公司披露https://www.theinformation.com/articles/little-known-startup-surged-past-scale-ai-without-investors)
推演到更广泛的数据市场,据估计,各大顶级实验室每年在向多家数据供应商采购专属数据集生成方面的支出高达10亿至100亿美元。随着定制数据集和强化学习(RL)环境被牢固确立为提升各学科模型性能的核心加速器,这些数字似乎还在持续攀升。
如果将大语言模型(LLM)的提升维度简化为:(1)获取相关的训练数据、(2)模型架构的演进,以及(3)算力的增长,那么这种向外部购买数据集的趋势在以下几个层面上就完全说得通了。
首先,过去几年里各种数据收集工作的叠加(例如Common Crawl对互联网数据的抓取、以及向出版商购买数据授权),已经加速消耗殆尽了所有现存的人类生成文本数据。
其次,由于实验室之间频繁的人才流动以及对研究的巨大投入,模型架构的重大突破往往会随着时间的推移而走向商品化(同质化)(例如Transformer架构),这使得架构本身很难再成为AI实验室手中可靠的护城河。
最后,算力瓶颈正越来越多地受到日益增长的模型推理(服务)需求的制约,而不是受限于训练模型所需的算力(Compute constraints are increasingly governed by growing demand to serve models rather than by the compute needed to train them)。
这种对数据的渴求,在编程、金融和电脑操作(Computer Use,指AI替代人类操作电脑)等当下最火热的应用领域表现得最为普遍。其背后的逻辑相当简单,而且这套打法似乎屡试不爽:在训练数据中加入更多精选的Python代码示例,大语言模型(LLM)很可能就会变成一个更优秀的软件工程师。
我越来越多地看到,针对用于训练生命科学基础模型的生物数据集,人们也在讨论相同的趋势。模型开发商们正在达成更多的授权协议,以获取与特定任务相关的数据。在训练数据中加入更多高质量的抗体亲和力测量数据(high-quality antibody affinity measurements),生成式蛋白质模型就很可能变成一个更优秀的蛋白质工程师(protein engineer)。
为了满足这一需求,人们也越来越关注提升收集这些生物数据集的速度和严谨性。自动化的生命科学机器人公司现在正将自己标榜为“数据代工厂”(Data Foundries),试图利用这个不断增长的市场,从而在数据收集、模型训练和模型评估之间构建一个闭环的飞轮。
在收集、买卖生物数据以及基于其进行模型训练的过程中,这些日益增长的机遇也引发了一些亟待思考的关键问题:为生物基础模型筛选和训练数据集的正确策略是什么?在用文本数据训练LLM的方法中,有哪些原则是可以移植的,又有哪些是无法通用的?
尽管前沿LLM与生物基础模型在宏观层面的市场表现上存在诸多平行相似之处,但它们在数据层面有着深刻且微妙的差异,非常值得深入探讨。
从数据的视角来看,LLM训练的历史打法—即一味将重心放在“数据规模”(Data Scale)上—将无法1:1复制到生物数据集上。相反,我认为筛选生物训练数据集需要更加谨慎,必须将数据质量置于数量之上,我们应当在陷入“规模至上”(Scalemaxxing)的陷阱之前,清晰地认识到这一点。
LLM预训练范式
The LLM pretraining paradigm
在现代大语言模型(LLM)预训练的早期阶段,行业内占据主导地位的共识是通过“规模化”(Scale)来提升模型性能。即更多的数据、更多的参数、更强的算力。这具体表现为整个领域耗费了大量时间去推演和列举“预训练缩放定律”(Scaling Laws)——即模型性能的提升幅度,是如何作为数据量、模型大小、算力等变量的函数(斜率)而变化的。关于这一主题,我们在之前的一篇我们的研究杂志《Dimension Research》文章中曾进行过深入的探讨。
注:Pretraining:The First Scaling Frontier
https://research.dimensioncap.com/p/pretraining-the-first-scaling-frontier
2020年,OpenAI的Kaplan等人发表了一些最初的预训练规模定律,表明在固定的计算预算下,以比数据(token数)更快的速度增加模型规模(参数数量)可以获得最优性能。2022年,DeepMind的Hoffmann等人发表了“Chinchilla”规模定律,指出基于Kaplan定律的模型训练不足,使用更多数据训练较小的模型(约每个参数对应20个token)可以获得更好的模型性能。大约两年后,Databricks的Sardana等人发表了对Chinchilla规模定律的修正,将推理需求纳入考量。在固定的计算预算下,为了达到期望的性能,使用越来越多的数据(每个参数对应约200至10,000个token)来训练较小的模型在经济上更为划算,因为对于客户而言,提供较小的模型进行服务最终成本更低(it’s more economical to train smaller models with increasingly more data(~200-10,000 tokens per parameter)to achieve a desired performance because serving smaller models is ultimately cheaper for customers)。
那段早期探索所带来的核心启示,恐怕早已深深烙印在许多AI研究者的脑海中:“数据越多,效果越好。”在当时,规模可以优先于数据质量,这是因为人类花了几个世纪的时间积累了一个庞大的高质量文本库(例如互联网片段、已发表的文献),而LLM几乎是免费继承了这份遗产。它们从一开始就能绕过数据质量与数量之间固有的权衡取舍(They were originally able to sidestep the canonical trade-off between data quality and quantity),因为这份继承下来的数据集在两个维度上都足够充沛,足以支撑起实用的模型性能。
研究人员可以一边利用规模效应,一边使用高层级的过滤手段(如去重、去污染、早期的质量分类器)来剔除低质量的样本。由于文本数据具有高度的冗余性,噪声也因此得到了进一步的容忍,这意味着在已有的庞大分布规模下,随机错误和前后矛盾可以被自动平均、相互抵消(so random errors and contradictions average out at the scale we already had)。而一旦模型能够自主生成高质量的合成数据,这种权衡的难度就更进一步降低了。
不过在过去两年中,整个领域已经开始看到这套打法的重新平衡。随着可用数据的消耗殆尽,对高质量数据集的重视,成为了预训练阶段另一个可供撬动的重要杠杆,研究人员也开始开发更加激进的数据优先级筛选工具(more aggressive data prioritization tools),以便在兼顾规模的同时实现这一目标:
1.基于重要性重采样的数据选择(Data Selection with Importance Resampling,DSIR):从一个庞大的数据集中,定义出一小部分高质量数据的子集。然后,在整个数据集更广泛的分布中去对这种“高质量特征”进行采样,从而把所有其他潜在的高质量样本都捞出来。
2.基于极大极小优化的领域重新配比(Domain Reweighting with Minimax Optimization,DoReMi):训练一些小模型,并让它们使用不同配比的数据类型(例如50%通识、25%编程、15%科学、10%推理轨迹),以此来寻找性能表现最佳的数据分布。一旦找到最优组合,再将这种分布配比放大应用到大模型上。
3.基于数据影响力模型的模型感知数据选择(Model-Aware Data Selection with Data Influence Models,MATES):训练一个小型的“数据影响力模型”(data influence model),它能够动态地为大模型预训练的各个阶段筛选出最有效的数据,从而优化模型接触数据的先后顺序。
4.质量分类器(Quality Classifiers)(FineWeb、DCLM、Nemotron-CC):训练独立的分类器模型,根据用户定义的指标对数据质量进行评分,并过滤掉任何达不到质量阈值的内容。更小但质量更高的数据集,其性能甚至可以超越质量参差不齐的更大规模数据集(例如,一个7B的DCLM模型在训练数据量减少约6倍的情况下,其性能表现与8B的Llama 3相当)。
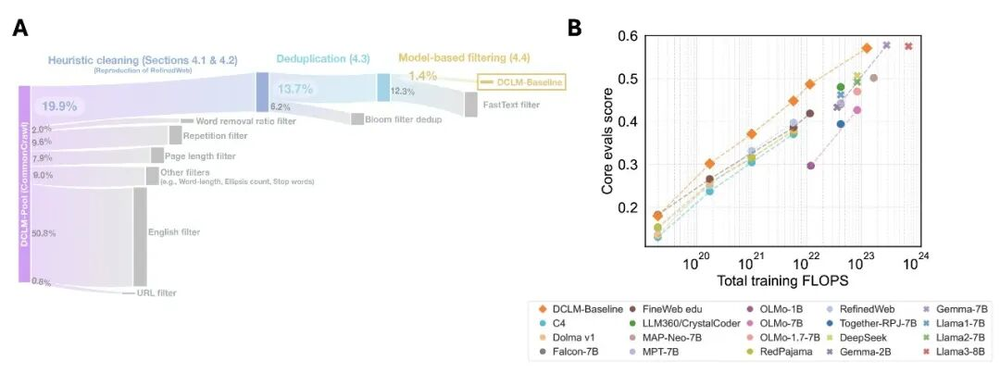
(A)利用质量分类器模型进行过滤,从原始的Common Crawl数据源中构建DCLM训练数据集。(B)基于过滤后的DCLM-Baseline数据集训练的模型,其性能超越了那些拥有更大训练预算(数据量)的模型。(插图改编自:https://arxiv.org/abs/2406.11794)
这些方法无一不彰显出数据质量对大语言模型(LLM)而言那重获新生的重要性。然而,它们之中的绝大多数都严重依赖一个前提:必须先有一个庞大的、定义清晰且高质量的现有语料库作为过滤的底盘。这对几年前的LLM来说不是问题,但如今,我们几乎已经把现有数据中能榨取的价值给“薅得一干二净”(milking nearly everything we can)了。
前沿实验室在特定应用数据上的巨额支出进一步证明,跨领域的高质量数据集已成为一个主要瓶颈(The huge spending from frontier labs on application-specific data is further evidence that quality datasets across domains have become a major bottleneck)。
值得庆幸的是,对于如何普遍应对更进一步的数据收集,我们手中已经有了一套相当不错的“通关秘籍”(recipe)。在许多我们关注的领域中,验证数据质量及其关联的基准测试(benchmarks)相对容易(例如:编写的代码能输出正确的结果;数学分析能推导出正确的闭式解;关于网球历史的报告符合事实且可供人类理解)。我们甚至可以对上述部分质量过滤工具进行改造并加以利用,从而在向外部采购新型数据时,能够分清轻重缓急。
然而遗憾的是,这套适用于LLMs的数据框架,并无法直接平移到生物学的世界里(this data framework for LLMs does not directly port to the world of biology)。
为什么生物学不适用这套做法
Why biology breaks this playbook
生物基础模型(Biological foundation models)正在众多子领域和应用场景中突飞猛进。在分子生物学领域的模型(Biomolecular models)正在实现生成全长、高亲和力单克隆抗体的计算机端(in silico)设计与生成。

在基因组基础模型(genomic foundation models)方面,AI已经开始预测变异效应(variant effects)并设计出逼真的DNA序列;而在组织层面的世界模型(tissue-level world models)开发上,科学家们正致力于预测空间蛋白质表达以及患者的药物反应(predict spatial protein expression and patient drug responses)。

这些成就部分可以追溯到过去几年深度学习和大型语言模型的突破。许多最初受文本模型启发的架构和规模直觉,随后被应用于公共的生物数据(Many initial architectures and scaling intuitions inspired by text models were subsequently applied to public biological data)。
但只有当数据经过良好整理时,这套做法才能真正奏效。典型的例子是AlphaFold,它展示了在蛋白质数据银行(Protein Data Bank,PDB)—一个经过数十年整理的、包含约20万个蛋白质结构的高质量实验数据集—上进行预训练,如何能在结构预测中带来惊人的性能。

但PDB更多是一个特例,而非普遍现象。与用于训练前沿LLM的文本数据截然不同,我们目前可以公开获取的生物数据要稀疏得多,并且饱受几种独特而显著的局限性困扰,这使得它们很难直接用于模型训练。因此,训练高性能的生物基础模型通常需要合成新的数据,而(生物)前沿实验室正开始探索类似于我们在大型语言模型领域看到的那种数据采购策略(data procurement strategy)来获取这些数据。
在这样一个世界里,针对给定的应用场景,生成什么样的训练数据才是最有效的?我们在生物数据收集上的底层哲学应该是什么(What should be our philosophy on biological data collection)?在生命科学中,当规模与质量之间的权衡取舍(trade-off)变得更加赤裸和尖锐时,我们该如何恰当地平衡两者?
回答这些问题,需要对何谓“高质量”给出一个更正式的定义,并理解现有生物数据集相比于文本训练数据究竟存在哪些局限。在这里,我将生物数据的“质量”定义为四个核心维度:(1)富含上下文信息(context-rich)、(2)干净(即低噪声且高可复现性)、(3)多样性,以及(4)为了探寻底层真实情况(ground truth)而量身定制(purpose-built)。
上下文的匮乏
Context Scarcity
想要与几乎整个人类有记录的历史规模相抗衡,确实是一件难事。据公开披露的指标,一些体量最大的大语言模型(LLM)在经过过滤后,其训练数据量依然高达数十万亿个Token。
但我在这里的核心论点,并不仅仅是“我们拥有的文本数据比生物数据多”。事实上,我们手头现存的生物数据量已经相当可观——包含了数十万亿个核苷酸碱基(nucleotide bases)、数亿条蛋白质序列、数亿个单细胞转录组图谱(single-cell transcriptomic profiles),以及数十万个通过实验测定的蛋白质结构。
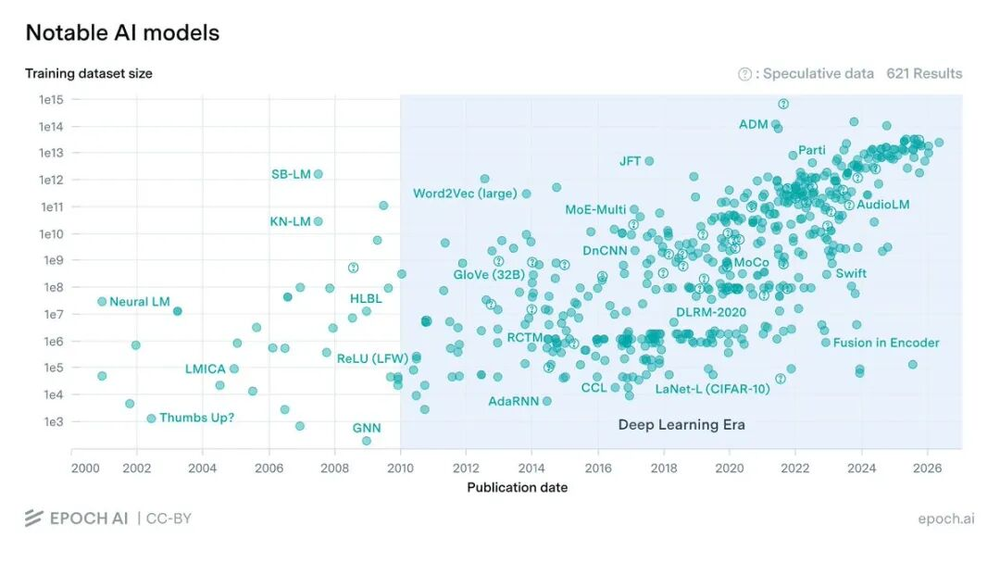
过去十年间,顶尖人工智能模型的训练数据集规模显著增长。(数据来源:https://epoch.ai/data/ai-models)
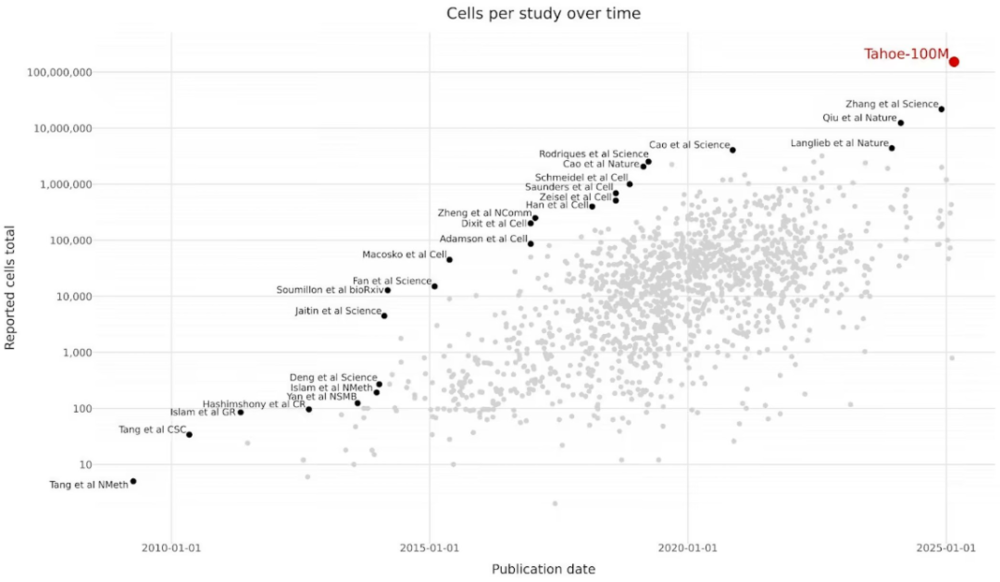
过去十年间,单细胞RNA测序数据集的大小(细胞数量)已显著增长。
https://huggingface.co/datasets/tahoebio/Tahoe-100M/blob/main/README.md
文本数据与生物数据之间更重要的区别在于上下文的匮乏。
对于文本数据,相关的上下文已经内置于语言之中—句子结构、文档组织、来回对话、逻辑性的话题解释。所有这些内容,无需额外进行繁重的标注,即可被人类和机器直接读取。这对于通过“预测下一个Token”(next-token prediction)来进行的自监督预训练尤为重要,在这种模式下,研究人员追求的是最大化每个Token所蕴含的学习信号。例如,当你将句子中的下一个单词遮蔽时,前面的词汇和句子结构就会自然而然地提供相关的上下文,以此来预测缺失的内容。
某些类型的生物数据,如蛋白质或DNA序列,具有与文本数据类似的内置上下文。例如,相邻的残基(residues)会限制某些特定基序(motif)中可能出现的氨基酸。我们甚至可以使用类似的掩码语言模型(masked language modeling)方法来学习生物序列中的这些模式,并预测缺失的氨基酸。
这种方法之所以如此有效,原因之一在于蛋白质和DNA序列中蕴含着天然的的演化上下文(evolutionary context)。我们可以通过多序列比对(Multiple Sequence Alignment,MSA)等技术将其提取出来,从而获得更强的预测能力。当你要预测的生物学特性恰好也是自然演化所筛选的特性时,这种方法会非常灵验。例如,一个残基之所以常常在演化中被保留(保守残基),通常是因为它与蛋白质的功能密切相关。因此,一个学到了广泛序列分布规律的模型,在理论上即使不针对特定的标签样本进行训练,也能预测出某种突变是否会导致功能的丧失(so a model that learns a broad sequence distribution could theoretically predict whether a mutation results in loss of function without training on a specific labeled example)。
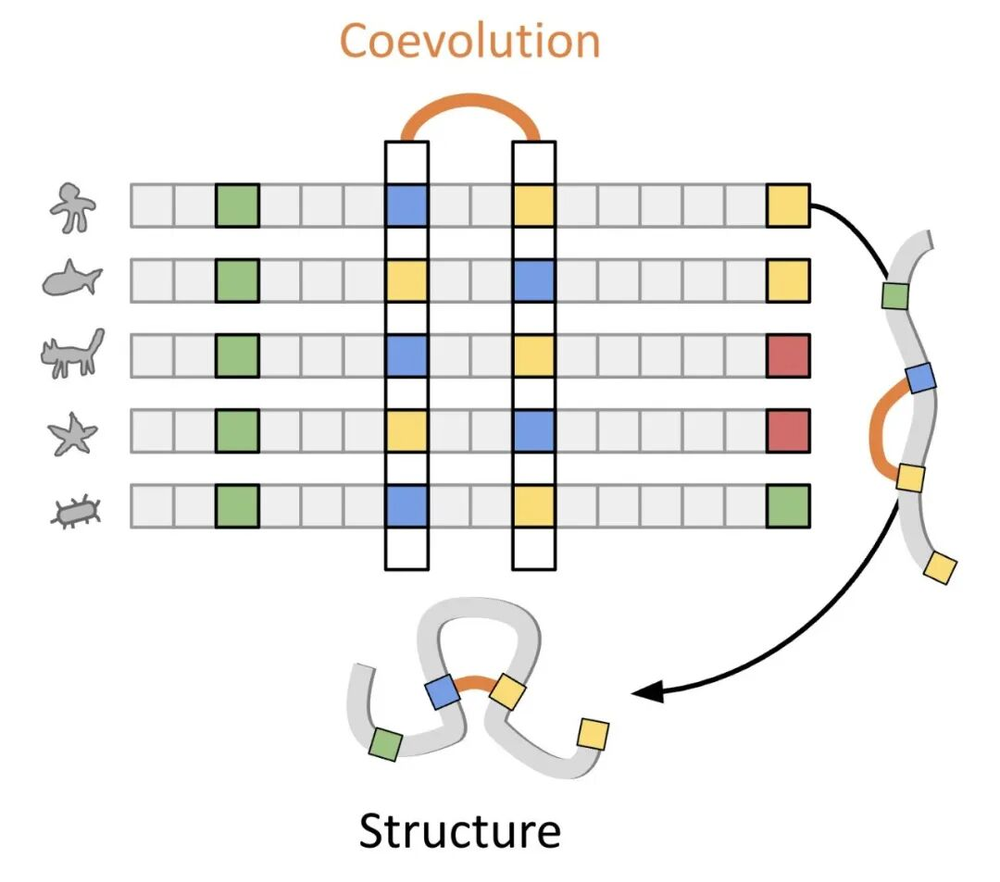
蛋白质序列的多序列比对可以识别出保守和共进化的残基,这些残基包含了关于蛋白质结构和功能的有用信息。
然而,演化信号(evolutionary signal)是有限的,如果我们将其作为唯一依赖的上下文,就面临着误读它的风险。例如,它无法完全体现自然演化中所有失败的尝试(例如胚胎致死性突变),也无法涵盖那些已经灭绝、或因为缺乏数据而未被充分采样(under-sampled)的生物体。此外,它还默认了一个前提,即“序列的保守性或协同变异(co-variation)就意味着重要性”,但这在科学上并不是绝对成立的。
或许最重要的一点在于,仅凭天然蛋白质序列带来的演化信号,并不能包含其他与药物研发(drug discovery)密切相关的实用上下文。比如:全新的蛋白质功能、与合成靶点之间的结合亲和力(binding affinities)、翻译后化学修饰(post-translational chemical modifications)、跨非靶点的选择性(selectivity across off-targets),或者是药物的可开发性(developability)特征。当我们在对细胞表型(cell phenotypes)等更复杂的生物学过程进行建模时,现有的数据集中,相关的上下文信息—如细胞基因型、环境条件以及实验方案层面的具体细节(protocol-level details)—就会变得更加匮乏。
上述所有缺失的上下文都存在于物理世界中,我们必须通过实验费力地测量它们,并使用这些测量结果来标注数据。此外,生成高质量、带标注的生物数据通常非常昂贵,并且对分析方法的设计和执行有着很高的要求,这可能成为非专业人士面临的重大障碍。对于湿实验科学家(wet-lab scientists)来说,成本、时间、质量和数量之间的权衡始终存在。
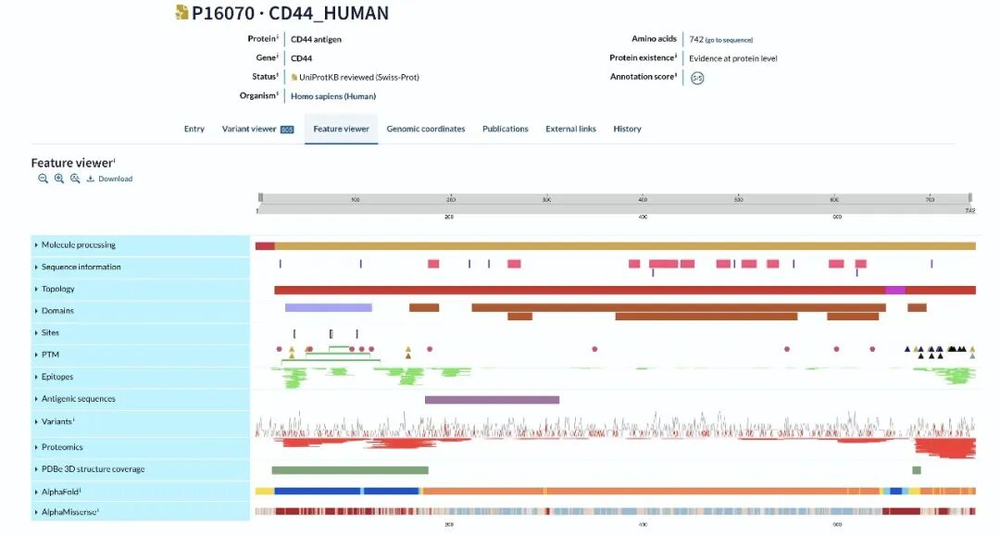
UniProt数据库中包含的、与生物基础模型训练数据相关的一小部分蛋白质序列注释。(https://www.uniprot.org/)
在这背后,还隐藏着另一个与之相关的底层问题。生物学的上下文完全受限于我们已经观察到和/或发现的内容,这从一开始就埋下了人类视角偏见的种子(Biological context is limited to whatever we’ve already observed and/or discovered,which bakes in bias from the start)。
因此,所有的生物数据集本质上都是不完整的(all biological datasets are incomplete)。人类可能还没有开发出测量那些缺失上下文的技术,甚至从一开始就根本无法理解哪些背景信息才是真正需要被标注的相关上下文。新的科学发现正在不断重塑我们对生物学的认知,并影响着我们优先去测定哪些上下文—比如,人类意识到实体瘤微生物组(solid tumor microbiomes)的多样性及其深远影响;发现RNA可以被糖基化(glycosylated)从而影响其定位与功能;亦或是人类基因组首次完成端到端(T2T)全序列测序,纠正了以往参考基因组中的错误,并引入了此前从未被纳入计算的约2亿个新碱基对。
分阶段预训练(Staged pretraining)是该领域试图兼顾两者优势、并部分弥补生物学上下文匮乏的方法。ESM系列模型就是一个极佳的范例:首先,在庞大的无标注蛋白质序列数据集上进行自监督预训练,利用其固有的演化信号生成强大的基础嵌入(base embeddings);随后,在较小但标注精细的数据集上进行序列微调(sequential fine-tuning),从而注入面向特定应用场景的性能。虽然这种方法比单独使用带标签的数据进行训练更高效,但其下游性能仍然严重依赖于用于微调的注释数据的质量和上下文完整性。
噪声
Noise
即使我们能够整理出一个完全标注、具备上下文感知能力的数据集,生物数据本质上仍然充满噪声,真实的训练信号可能很难被干净地提取出来。
你可能会想,从噪声中寻找信号正是深度学习擅长的事情。从很多方面来说,你是对的。在混乱的网络数据上训练的模型,仍然能学习到流畅的语法和广泛的世界知识,因为文本是冗余的,其噪声大多是随机的且不相关,因此大量噪声会在规模扩大时被平均掉了。
生物噪声则复杂得多。它有多种形式,而且往往无法解耦(deconvolve)。这一点很重要,因为某些这种“噪声”实际上是在扮演有用信号的角色,我们希望可靠地捕捉并学习它(some of this“noise”is actually moonlighting as useful signal that we want to reliably capture and learn from)。例如,生命科学研究中的一个核心挑战是:在接纳生物“噪声”(如患者间的异质性或肿瘤中的亚克隆多样性)的同时,减少技术噪声(technical noise)(如低计数测量中的散粒噪声或动态范围有限的代理读数)和系统噪声(systematic noise)(如用户间的批次效应或不同实验室累积的方案差异)。
分离这些主要的变异来源,使我们能够更真实地代表生物样本的多样性,这对于现实世界的药物发现任务(如预测不同患者群体的治疗反应)至关重要。举个具体的例子,我几年前写过一篇综述文章,主题正是关于使用类器官真实模拟人类肿瘤样本这一具体问题。
问题在于,我们在生物学中的所有测量都会引入技术噪声和系统噪声,其中一些我们了解得较少。通常,这些噪声信号的强度足以掩盖实际的生物信号,使其极易受到意外偏差的影响。幸运的是,技术噪声在规模扩大时更容易被平均掉,但系统噪声则不然。模型最终可能会将某些细胞表型与产生数据的实验室、使用的仪器、甚至样本是在孔板边缘还是中心相关联,而不是学习真正区分它们的底层生物学特性。
例如,为训练生物基础模型而收集足够的数据,通常意味着将不同实验室的相关数据集整理到一个集中的公共数据库中(例如,用于细胞表达数据的CELLxGENE,用于分子生物活性的ChEMBL)。由于系统噪声的存在,存在相当大的批次效应,这些效应会被融入模型对生物学的表征中;如果我们不小心,更多的数据可能会使这个问题更加突出。
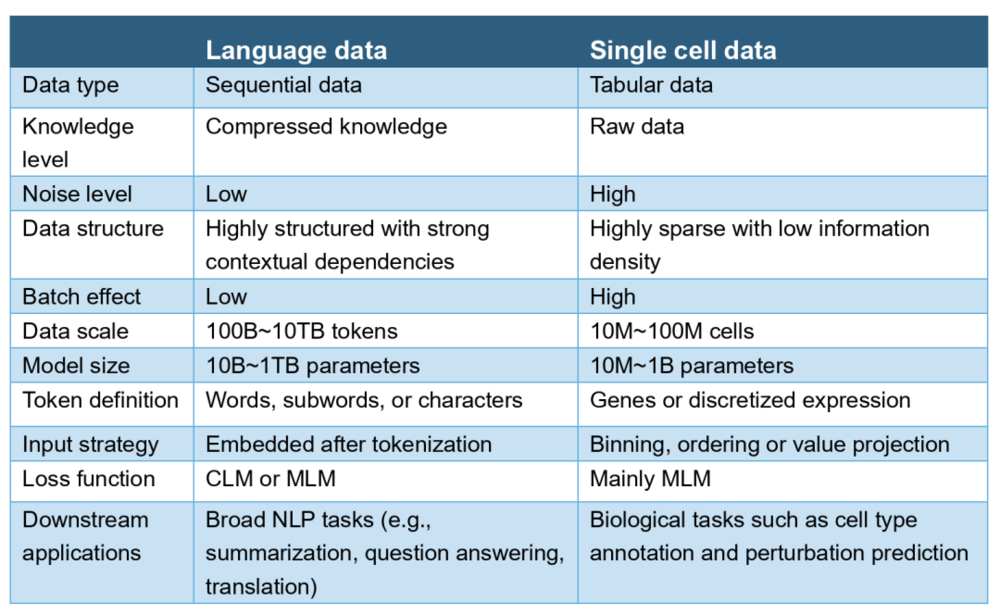
语言数据与单细胞数据的比较特征。MLM:掩码语言建模;CLM:因果语言建模;NLP:自然语言处理。来源:
https://www.biorxiv.org/content/10.64898/2025.12.19.695371v1
甚至有一项研究发现,他们仅凭ChEMBL数据库中的结构数据,就能训练出一个分类器模型来预测是由哪位化学家合成了该分子。这是因为不同的实验室往往会围绕特定的特征支架(scaffolds)开展研究,从而在它们制造的分子中留下了可被识别的独特印记。作者随后表明,仅靠“化学家身份”这一个变量,对分子生物活性的预测准确度,就几乎与包含完整结构信息的模型不相上下。这有力地表明,模型完全可以学会通过这种方式来“作弊”,从而在没有真正理解核心“结构-活性关系”(SAR,Structure-Activity Relationship)的情况下,就能猜出分子的活性。

相比之下,用于生成和收集文本数据的技术近乎完美。我们可以聘请世界专家就新的主题领域撰写报告,并使用网络爬虫工具提取现有的数字化文本。我们甚至拥有像自动更正和拼写检查这样无处不在的工具,能在数据集进入训练运行之前就实时对其去噪。此外,我上面提到的文本数据筛选工具依赖于已经包含高质量数据的现有大型数据集,而这在生物学领域并不总是成立。
多样性
Diversity
当面对其训练数据集分布内(in-distribution)的任务时,目前的AI模型表现确实异常出色;然而,一旦要求模型泛化到这种分布之外,其性能就会显著下降。因此,各领域的一个主要工作方向就是扩大训练数据的多样性,从而将尽可能多的相关样本纳入到分布之内。
但数据多样性并不仅仅指一件事。。它(粗略地)可以由两个主要的子维度来代表:样式多样性(stylistic diversity)和语义多样性(semantic diversity)。
对于文本数据而言,样式多样性是相同的基础内容以不同的方式表达—同一事实以随意、正式、跨语言、冗长或简洁的方式呈现。另一方面,语义多样性描述的是基础内容扩展了数据集所代表的核心信息—不同主题下的不同事实(when the underlying content expands the core information represented by the dataset—different facts across different topics)。前者能确保模型是可引导的(steerable),不会死板地陷于某一种文章写作风格或某一种特定的问题解法中;而后者则确保了模型能够在更广泛的子领域中都有良好的表现。
这些分类也大致适用于生物数据:
生物学中的样式多样性:类似于用不同的方式去测定相同的生物学输出—比如在两个不同的实验室里测定相同的结合亲和力,或者分别利用X射线晶体学(crystallography)和冷冻电镜(cryo-EM)来测定同一种蛋白质结构。
生物学中的语义多样性:则通过测定完全全新的生物学特性或标注来扩展数据分布(Semantic diversity expands the data distribution by measuring completely new biology or annotations)—比如全新的蛋白质序列或功能属性(new protein sequences or functional attributes)。
与前文关于上下文匮乏的论点类似,我们目前数据集中所具备的生物学多样性,远比文本数据要匮乏得多。如果处理不当,这两个子维度将会给生物模型的推进带来各自独特的风险。
例如,生物学中的样式多样性是一把双刃剑。如果处理得当,增加跨实验方案(protocols)或跨技术收集的数据多样性,可能会提供有益的信号,从而让底层模型对变异具备更强的鲁棒性。但如果我们不够谨慎,样式多样性就会与生物学噪声形成特有的交织缠绕,并冒着引入批次效应(batch effects)的风险,这反而会削弱模型的泛化能力。
而由于以下几个原因,现有数据集中的语义多样性同样一直处于匮乏状态:
我们许多体量最大的生物数据集都是从自然界存在的生物样本中挖掘出来的(Many of our largest biological datasets are mined from biological samples that occur naturally)(例如UniProt中的蛋白质序列、PDB中的蛋白质结构、GenBank中的基因组)。由于实际操作的原因,这些生物数据集仍然只代表了自然存在的一小部分,而自然存在本身又只是所有可能客观存在的生物多样性中非常小的一部分(these datasets still only represent a small slice of what naturally exists,which in itself is a very small slice of all possible biological diversity)。自然演化受限于幸存者偏差(survivorship bias),并未对所有可能的设计进行采样。
这在生成式蛋白质设计(generative protein design)等领域演变成了一种制约,因为许多我们想要研发的药物分子,其结构可能与自然界中发生过的任何东西都毫无相似之处(many drugs we want to develop may look nothing like what has occurred naturally)。
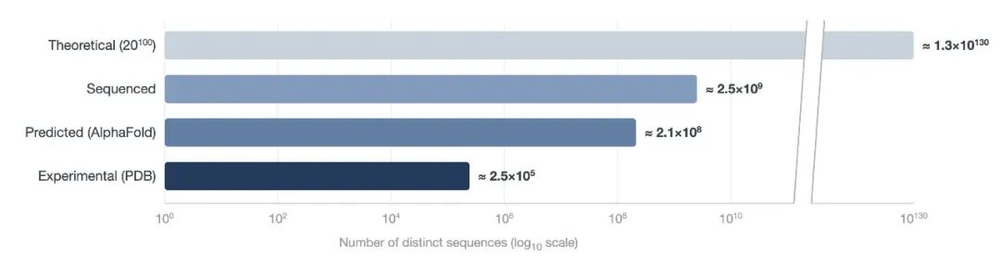
此图直观地展示了与总可能序列空间相比,经实验解出和计算预测的蛋白质结构以及已测序蛋白质的数量相对较少。其中,“已测序”类别是根据UniProt和宏基因组目录估算得出的。“理论”类别以100个氨基酸的序列为边界,代表了使用天然氨基酸所能达到的总搜索空间的上限。
除了自然演化带来的偏差(evolutionary bias)之外,我们目前生物数据集的多样性还受到科学兴趣,以及哪些项目能获得资金资助的严重偏置(scientific interest and what’s fundable)。人们在筛选和治理数据时,往往是为了试图回答非常具体的生物学假设,而不是为了给未来训练机器学习模型做储备(Data is often curated in an attempt to answer very specific biological hypotheses,rather than to future-proof for training ML models)。
合成数据生成(Synthetic data generation)是目前正在探索的、用以弥补数据多样性不足的领域之一。其理念是,当模型的输出达到高质量时,这些输出就可以用于生成其自身的训练数据。Meta公司最近的一项研究发现,用合成文本补充真实训练数据,其效果可以匹配甚至提升仅在真实数据上训练的大型语言模型的性能。

但这是一条危险的道路。合成数据的质量必须很高,并且过度依赖合成数据集可能导致模型坍塌,即性能显著下降。
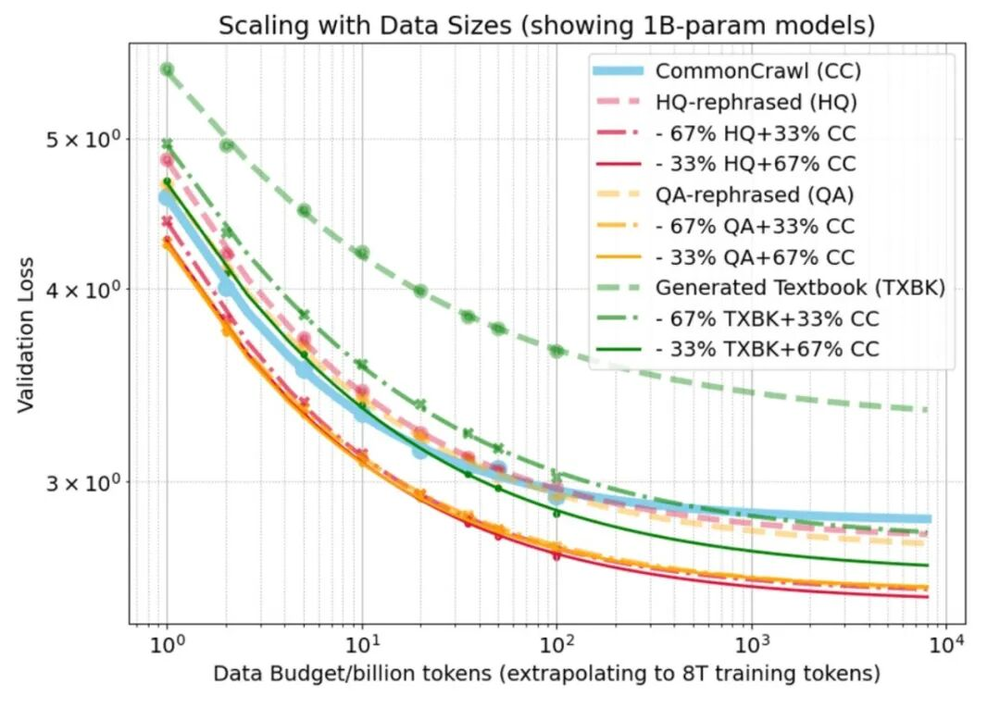
验证损失(Validation loss)与训练数据预算(数据量)的关系,对比了真实网络数据(Common Crawl)与两种合成数据:(1)将真实网络数据改写(Rephrased)为更高质量格式的文本(HQ和QA),以及(2)从头开始生成的文本(Generated Textbook)。图中展示了每种数据集单独训练以及与真实数据按不同比例混合训练的结果。结果表明,基于真实文本改写的合成数据,其表现与真实数据基线持平甚至更优;而从头开始生成的文本,其所占权重越高,模型表现就越差。这说明合成数据在锚定(依附于)真实数据时是有帮助的,但如果脱离了真实数据,则会导致性能退化。(https://arxiv.org/abs/2510.01631v1)
正如你所料,合成文本与合成生物数据当前的效益开始出现分歧。
对于写作等任务,合成生成的文本可以提供有价值的训练信号,并且只要读起来大致合理,它在本质上就是“正确”的。人类(甚至大型语言模型本身)通常仅通过阅读就能很容易地判断合成文本的质量是好是坏。
而对于验证本身就极为困难的生物学来说,合成生物数据只有在与实验室的实验结果相符时才是“正确”且有意义的。我们可以轻松地使用生成模型设计出数百万个看似合理的蛋白质结构,但它们的真实功能或结合亲和力在实验室中实际测量之前仍然是未知的。任何生成的合成标签都只会继承生成它们的模型的偏差或批次效应;如果没有持续的验证,你就有可能是在使用有缺陷模型的伪影(artifacts)而非真实的生物信号上进行训练。
当然,在一些非常特定的生物学应用中,我们已经看到了合成数据生成所取得的早期成功。例如,AlphaFold 2在训练中引入了“自蒸馏”(self-distilled)的合成数据,这些数据是基于PDB数据库中并不存在的未知序列生成的,从而成功提升了模型在这片外扩序列空间上的预测性能。这一策略之所以能奏效,是因为AlphaFold 2的结构预测结果已经达到了足以用于训练的“质量门槛”(quality bar),并且在需要时,人们有一套清晰明确的方法可以在湿实验室(wet lab)中对这些结构预测进行交叉验证。
底层真实
Ground truth
这最后一个质量指标是直接建立在前三个指标之上的。即便拥有了丰富的上下文、干净的信号和多样化的数据,生物学在本质上依然极其复杂,而且验证其底层真实(ground truth)往往异常困难。
在任何领域中,可靠的输出验证都是完成数据反馈闭环(data feedback loop)、并通过强化学习(reinforcement learning)来提升模型性能的核心机制。正如我开头所提到的,像编程这样的领域往往原生自带验证机制(fields like coding often have verification baked in),这也是为什么编程智能体(coding agents)在过去一年的性能表现呈指数级增长的核心原因。
然而在生物学中,底层真实情况必须通过实验手段来测定,这比执行一段代码要昂贵、复杂且耗时得多。这些测量结果也常常是我们试图记录的实际信号的代理(例如,用光子来测量荧光标记蛋白质的表达),而且这些信号通常只在很小的动态范围窗口内呈线性(these signals are usually only linear in small windows of dynamic range)。同时,在药物研发的应用场景中,我们对于这些测量结果对“人类临床疗效”的正向预测值(positive predictive value)到底有多少,也缺乏真正透彻的理解。
这些局限性促使该领域创造性地思考生物数据的规模化应该是什么样子(to think creatively about what scaling biological data should look like)。最近一篇关于黑箱数据扩展的论文,为未来的框架提出了几个令人信服的观点。在该论文中,作者认为,我们不能依赖数据集的人类可读性或启发式方法,来达到训练高性能生物基础模型所需的规模。相反,数据源需要针对“机器消费而非人类直觉”进行优化(data sources need to be optimized for“machine consumption rather than human intuition”)。

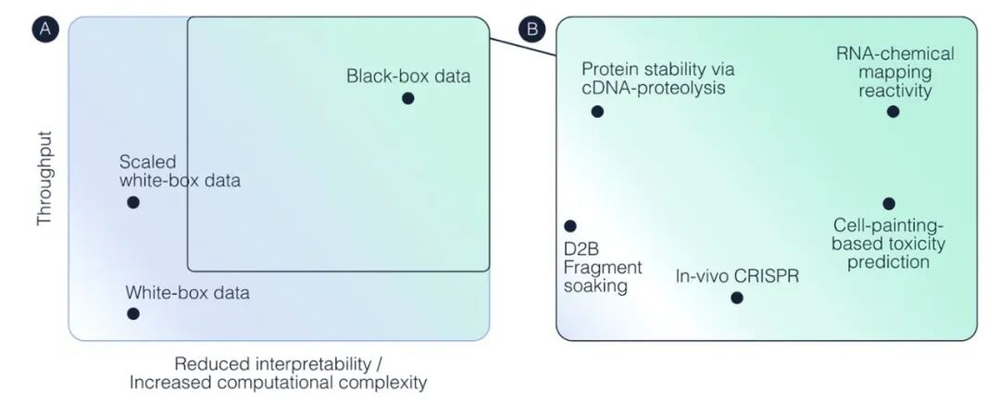
白盒数据与黑盒数据的对比。黑盒数据通过提升计算处理的复杂度,以牺牲可解释性为代价,换取了更高的数据吞吐量。
https://pubs.rsc.org/en/content/articlelanding/2026/sc/d6sc01189f
尽管我在这篇文章中采用了这样的叙述框架,但我认为我们最终仍会逐渐接纳这一(黑盒规模化)底层哲学。不过我的核心论点是:在此之前,我们必须保持平衡,同样将重点放在建立一套能够最大化数据质量与生物学理解的“行动指南”(playbook)上。
一个极具代表性的例子,便是近年来兴起的、基于转录组学数据训练以预测细胞对扰动(perturbations)反应的“虚拟细胞”(virtual cell)模型。
这些模型的训练数据通常局限在一个极窄的永生化癌细胞系(immortalized cancer cell lines)范围内,向特定的组织类型严重倾斜,且仅仅捕捉了单一点的时间戳快照表达数据;不仅如此,它们还将来自不同实验方案、具备不同测序深度和噪声特征的单细胞测序结果生硬地拼接在了一起。尽管这些数据集涵盖了高达近1亿个细胞的庞大体量,但多项独立的基准测试表明,在与药物研发最紧密相关的下游任务中,这些模型的表现依然难以超越一些简单得多的传统方法。


我的目的绝非粗暴地将整个子领域全盘否定。事实上,不同的虚拟细胞模型所使用的数据类型各不相同,且在推动这些模型向前发展的过程中,已经沉淀了大量重要的基础设施与前沿探索。但这个例子是一个有益的提醒:数据数量的规模化(scaling)往往会跑赢数据质量的规模化,而数据中的偏见(biases)与上下文的断层(gaps in context),常常会以模型自身无法察觉的方式死死地烙印在模型之中。因此,如果没有适当的验证、生物学洞察力或质量控制,过快地进行规模化可能导致我们在浑然不觉的情况下,将黑盒模型向着“错误的方向”规模化。而且,考虑到生物学中验证循环的速度之慢,我们可能需要很长时间才能意识到这一点。
这对模型开发者和数据生成者的意义
What this means for model developers and data generators
上述四个方面的分析表明,与处理文本数据相比,收集和训练生物数据需要一套经过修正的方法。只追求规模而不密切关注质量,是无法实现目标的。我们需要在以下三者之间建立一个紧密的闭环:生成高质量、与任务相关的训练数据的方法;能够准确验证这些数据的评估体系;以及在生命科学领域真实任务中接受测试的模型(a tight loop between methods for generating high-quality,task-relevant training data,evals that can accurately validate that data,and models tested against real-world tasks across the life sciences)。理想情况下,这三者应结合成一个足够快的飞轮,使每一轮验证都能为下一轮的数据设计(next round of data design)提供信息。
这说起来容易做起来难。在模型开发和数据生成这两方面—而且很多团队两者都做—有许多优秀的团队正朝着这些目标努力。以下是我认为对两个阵营而言最具机遇的领域。
面向模型开发者
For model developers
正如我们在前沿大语言模型(LLM)中所看到的,生物模型开发者对新型训练数据集的渴望也正变得愈发强烈,以期提升模型在基准测试(benchmarks)中的表现。随着我们步入这一市场动态中,开发者应当重点思考三个日益重要的问题:(1)如何评估数据提供方的数据质量?(2)如何验证这些数据对现实世界基准测试的实际效果?(3)在资源有限的情况下,何时进行规模化扩张(scale)?
(1)在未来的这一趋势中,数据采购将变得更像是一个科学同行评议(peer-review)的过程,而不仅仅是传统的资本与资产的简单交换。你购买了哪些数据固然重要,但关于这些数据是如何被收集和标注的元数据(metadata)也同样关键。实验方案级别的细节(protocol-level details)、一致性指标(consistency metrics)、批次结构(batch structures)以及技术重复(replicates)的处理方式,共同构成了一个更完整的数据包背景故事。这对于评估数据质量以及训练更可靠、更具上下文感知能力(context-aware)的模型至关重要。
鉴于生物学的极度复杂性,以及需要在高维坐标轴(high-dimensional axes)上对质量进行度量,目前用于文本数据的质量分类器、批次校正器和过滤工具,很可能无法直接移植到生物模型中。因此,开发生物学特异性的工具集应当成为模型开发者(甚至是介于模型开发者和数据生成方之间的第三方服务商)的一项核心任务,唯有如此才能实现这一流程的规模化。
我们已经可以在实际生产中看到这样的例子。例如scVI,这款最初专门为单细胞RNA测序(scRNA-seq)数据进行批次校正而开发的工具,现在也正在被用作一种质量过滤器,用来检测某个数据集中的信号究竟是由用户的批次效应驱动的,还是由底层的基本生物学规律驱动的。
(2)模型开发者还需要通过评估来验证性能,但鉴于上文讨论的诸多问题,为生物模型构建稳健的评估体系本身就是一项挑战。此外,留出的测试集通常与训练数据存在相同的批次效应,这意味着基准性能往往反映的是学习到的"伪影",而非学习到的生物学特性(which means benchmark performance often reflects learned artifacts rather than learned biology)。

如今最常用的评测体系在设计上都在尽力去匹配模型的复杂度。例如,一个抗体设计模型很可能拥有一整套涵盖亲和力(affinity)、选择性(selectivity)、功能性(functionality)、聚集倾向(aggregation)等多个维度的基准测试。然而,随着模型的输出变得越来越复杂,验证这些输出所需的手段也变得越来越定制化(bespoke),随之而来的便是验证时间和成本的急剧攀升。走到这一步,可能会有越来越多的模型开发者选择在内部自建湿实验室(wet labs),或者与数据生成方建立更深度的战略合作,从而在一个更受控、更垂直一体化的飞轮(verticalized flywheel)中缩短验证周期。
我们必须开始将验证环节推向尽可能贴近现实世界应用的前沿(pushing validation as close to the real-world applications as possible)。在LLM领域,Andon Labs就是一个极具启发性的范例:他们让AI智能体去实际运营Anthropic公司内部的真实自动售货机业务,以及位于旧金山的一家实体店。


andonlabs.com
在这里,对智能体成功与否的评判完全取决于最终的真实经济效益—这种硬指标可比那些人为设计的虚拟基准测试要难刷得多(where success is graded on actual economic outcomes that are much harder to game than an artificial benchmark)。
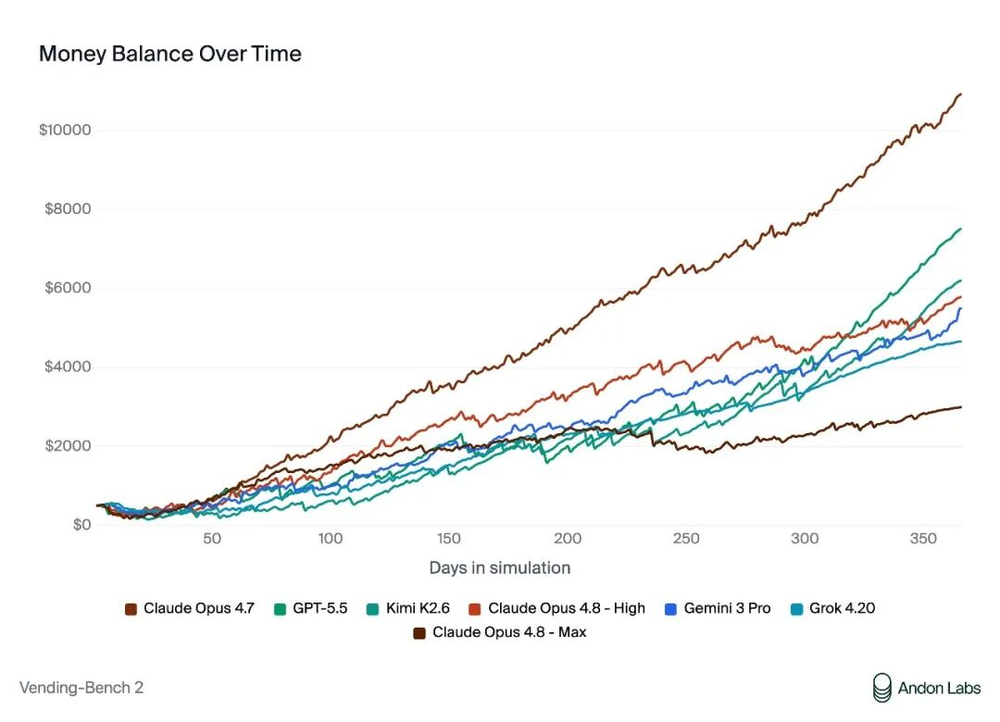
以下是来自Andon Labs的Vending-Bench 2评估结果的翻译。人工智能模型被给予500美元的起始资金,来自主管理一个虚拟的自动售货机业务。图表展示了每个模型随时间的现金余额变化。(https://andonlabs.com/evals/vending-bench-2)
对于生物模型而言,与此等价的闭环很可能就是药物研发流程本身。模型的性能最终将由其输出能否在临床前(preclinical)和临床(clinical)读出结果中存活下来而决定。这里的核心挑战在于“周期时间”:相比于自动售货机或零售店每天或每周就能给出的经济效益反馈,药物研发往往需要耗费数年的时间。尽管如此,两者的核心初衷是一致的,而且将“临床成功”作为终极衡量标准的终点线(goalposts)从未改变。
(3)设计一个能完美解决上述所有四个问题的分析方法或数据收集策略,很可能是不可能的。我们受到有限预算、短暂时间线和有限技术的约束(finite budgets,short timelines,and limited technologies)。在这样的环境下,开发者需要理解,何时是推进数据收集方案的正确时机,以及何时应该扩大规模(developers need to understand when the right moment is to advance data collection protocols,and when to scale)。
这里没有唯一的正确答案,但只有当开发者确信数据对于下游任务确实具有代表性时,规模化才应该开始。例如,空间转录组学(spatial transcriptomics)比单纯的单细胞RNA测序转录本计数提供了更多的上下文信息,但空间数据的收集比单细胞RNA测序要昂贵和耗时得多。如果你的主要应用场景依赖于理解组织内细胞类型之间的定位,那么这种额外的上下文信息就是一个值得优先考虑的权衡。
其他几个重要的考量因素包括:(1)每个样本中存在多少学习信号;(2)明确究竟是哪些噪声源在数据收集中占主导地位,以及如何最好地去缓解它们;(3)获取数据所需的时间和成本;(4)数据收集完成后,你能够多快对其进行实验验证;(5)预测错误的代价有多大,以及一旦出错,这项规模化投资的“可逆性”有多高(是否能及时止损)。
让情况变得更加复杂的是,不同类型的数据毫无疑问需要截然不同的规模。在某些应用中,基于100例带有明确临床预后(clinical outcomes)标注的癌症患者病例进行训练,其效果可能远好于基于10,000例仅带有“代理生物标志物”(surrogate biomarkers)标注的病例——因为这些代理指标与真正的临床终点(true endpoint)之间往往只有微弱的相关性。
这里存在着一个真正的“先有鸡还是先有蛋”的悖论。不进行规模化,我们可能就无法学到正确的生物学规律;但规模化跑得太快,又面临着将模型死死锚定在错误或不完整的生物学认知上的风险。行业内一个残酷的真相是:在真正训练出模型并开始摸索之前,你根本无法确切知道自己的数据到底有多好(you don’t know how good your data actually is until you train a model and start to find out)。这种内在矛盾张力警示我们,必须从一开始就将“质量”与“规模”放在同等重要的位置上通盘考虑,而不是把质量和生物学洞察仅仅当成是在粗放规模化之后、可以简单靠后期过滤就能解决的事情。
面向数据生成者
For data generators
生物基础模型正变得越来越复杂,将需要比公开可用或模型开发者内部能生成的更多数据。即使是地球上最有价值的制药公司—拥有数十年数据积累的礼来公司—也已经建立了一个数据共享平台,以满足开发药物发现AI模型所需的高容量、多样性和高质量数据。
数据生成者有多种类型:(1)拥有高通量筛选平台、能产生独特生物学见解的生物技术公司;(2)致力于规模化有价值实验流程的自动化/机器人公司;(3)多年来为客户精炼数据收集的合同研究组织(CRO);(4)甚至是专门为模型开发者优化数据收集的新公司类别。
我的论点是,那些越来越专注于高质量数据、并完善向模型开发者交付数据流程的参与者,将获得最大的成功(those players who increasingly focus on high-quality data and who perfect the data handoff to model developers will be the most successful)。他们为社区提供的价值主张是多方面的,我将特别关注其中三点。
1.这是目前最直接、也最简单的价值主张。这种独特优势可以归功于一种全新的筛选平台,或者是将一个已被验证的平台推向了一个全新的治疗生态位(therapeutic niche)。然而,这里最容易被外界忽视的环节,其实是对数据集进行穷尽式的标注(exhaustive annotation),以及落地这一标注所需的基础设施——这涵盖了从特定数据属性到精细至“究竟是哪位操作员在运行这一实验”的每一个细节。平台、数据以及这些精细的标注,共同构成了公司的护城河(moat)。
如果没有对注释和必要基础设施的大量投入,这种价值主张是短暂的(Without heavy investment in annotation and the required infrastructure,this value proposition is fleeting)。分析方法—即使是复杂的—往往会随着时间被商品化,参与者可能会被新技术或他人技术的更新所超越(Assays—even complex ones—are often commoditized over time,and players can be leapfrogged by new technologies or updates to the techniques by others)。这些参与者也可能在早期凭借价格和数据获取速度获胜,但存在与竞争对手陷入低价竞争的很高风险。
2.随着模型开发者在对生物数据机会进行尽职调查时变得越来越精明,生成高质量数据的声誉会为这种护城河增添价值。那些拥有可记录、可追溯的实验方案、机器可读的元数据、恰当的批次控制、充分的重复测量以及一致注释质量的数据生成者,会成为该领域值得信赖的来源,这直接转化为定价能力。无论底层生物数据多么有趣或独特,糟糕的数据质量和缺乏组织都会产生复合效应,而每一项质量改进都能提供更大的杠杆作用。
3.第三个、也是最成熟的价值主张,开始彻底改变数据生成者与模型开发者之间的关系形态。如今,CRO的默认业务模式绝大多数是被动响应式(reactive)的:他们接收客户发来的实验需求,运行实验、打包并交付数据,然后收取服务费。这种商业模式往往被“哪家供应商能以最便宜的价格、在最短的时间内满足最低的质量标规”所驱动—这又是一场逐底竞争。
事实上,数据生成者对当前模型的局限性有着独特的洞察,而这些局限性往往是开发者自己都浑然不知的。当数据生成者开始采取以下两项举措时,这种洞察就将开始解锁全新的价值:
走向产业链上游(move upstream):主动去审视和界定哪些数据集最有可能提升模型的评测效果(evals)。
构建专属的评测体系:亲自制定评测基准,以在特定任务中证明自身数据的核心价值。
随着高质量数据成为模型开发者越来越强的护城河,这种关系将从简单的“数据供应商关系”转变为深度绑定的商业战略合作伙伴关系,这无疑将带来更大且更持久的经济利益(lasting economics)。同时,我推测模型开发者未来会更倾向于获得数据集的独家授权许可(exclusive licenses),这使得从双端共同培养这种更紧密的共生关系变得尤为重要。
结语
Parting thoughts
无论哪个子领域,训练数据始终是人工智能进步的核心。对于具有自身独特考量的生物基础模型而言尤其如此(biological foundation models that come with their own unique considerations)。
显然,我们如何规模化并训练生物基础模型,不会有一个“一刀切”的方法,这应该受到行业和学界的欢迎。由于生物学极其恐怖的复杂性,每一个子领域都需要定制属于自己的模型架构、分词/表征策略(tokenization strategies),以及去仔细权衡数据质量与数据规模之间究竟该如何达成精准的平衡。
我在此处所阐述的任何观点,都不是在反对“规模化”(Scale),而是在为“高质量、多样化的数据”正名。如果只有规模而没有质量,或者只有质量而没有规模,我们都无法在生物学数字化(digitizing biology)的道路上取得实质性的进展。在一个仅仅对我们“当前已知的生物学认知”进行盲目粗放规模化的世界里,我们将丢失掉那些人类甚至尚未发现、或还不知道该如何去测定的关键上下文背景;而在一个过度沉迷于把数据集打磨到毫无瑕疵的世界里,我们又会失去利用有意义的模型去推进当下药物研发的能力。不要忘记,我们的终极目标,依然是尽快将有价值的资产(药物/疗法)传递到患者手中(The end goal is still translating meaningful assets to patients as soon as possible)。
我经常听到“数量本身就是一种质量”(quantity is its own form of quality)这种说法,并且我大体上同意这个观点,但我的意思是,这句话在生物学领域成立的前提是,数据首先要具有足够高的质量。随着该领域开始为地球上一些最棘手的问题构建更强大的生物模型,数据瓶颈的解决将不在于只关注“我们能收集多少数据?”,而在于“我们首先应该收集哪些数据?”。
原文链接:
https://research.dimensioncap.com/p/on-training-data-for-bio-ai-models